C. elegans releases
Release Notes
The 20210121 release includes genotypes from whole-genome sequences and reduced representation (RAD) sequencing. Genotypes are compared for concordance, and strains that are 99.97% identical to each other are grouped into isotypes. One strain within each isotype is the reference strain for that isotype. To look up isotype assignment, see Isotype List. All isotype reference strains are available on CeNDR.
- Strains: 1378
- WGS strains: 1238
- Isotypes: 540
- Genome: WS276
Datasets
| Dataset | Description | Download |
|---|---|---|
| Strain Data | Includes strain, isotype, location information, and more. | 20210121_c_elegans_strain_data.csv |
| Strain Issues | This link contains all strain issues up to this release | |
| Alignment Data |
Alignment data are stored as BAM files, which are binary representations of the Sequence Alignment/Map format. The specifications for these file formats continue to develop. Current specifications for BAM and VCF can be found at hts-specs. |
Alignment data are only provided for the latest release version. |
| Variant Data | Strain-level variant information is stored in the VCF and genomic VCF format. The gVCF format contains information for every base regardless of whether a variant is present or not and is suitable for compiling and joint calling variants across a custom strain set. These files were produced by GCTA. | Individual strain genomic variant files are only provided for the latest release version. |
| Soft-Filtered Variants |
Variant information is stored in the VCF format, which is a tab delimited format for storing variant calls and individual genotypes. It is able to store all variant calls from single nucleotide variants to insertions and deletions (~50 bp).
The soft-filtered VCF includes all variants and annotations called by the GATK pipeline.
The QC status of each variant (INFO field= The specifications for these file formats continue to develop. Current specifications for BAM and VCF can be found at hts-specs. |
All Strains
WI.20210121.soft-filter.vcf.gz WI.20210121.soft-filter.vcf.gz.tbi Isotypes WI.20210121.soft-filter.isotype.vcf.gz WI.20210121.soft-filter.isotype.vcf.gz.tbi |
| Hard-Filtered Variants |
Variant information is stored in the VCF format, which is a tab delimited format for storing variant calls and individual genotypes. It is able to store all variant calls from single nucleotide variants to insertions and deletions (~50 bp).
The hard-filtered VCF includes only high-quality variants after all variants and genotypes with a failed QC status are removed.
To obtain vcf for a single or a subset of strains, use The specifications for these file formats continue to develop. Current specifications for BAM and VCF can be found at hts-specs. |
All Strains
WI.20210121.hard-filter.vcf.gz WI.20210121.hard-filter.vcf.gz.tbi Isotypes WI.20210121.hard-filter.isotype.vcf.gz WI.20210121.hard-filter.isotype.vcf.gz.tbi |
| Imputed Variants |
Variant information is stored in the VCF format, which is a tab delimited format for storing variant calls and individual genotypes. It is able to store all variant calls from single nucleotide variants to insertions and deletions (~50 bp). The imputed VCF includes all the variants from the hard-filtered Isotype VCF, but all missing genotypes have been imputed using Beagle v5.1. The specifications for these file formats continue to develop. Current specifications for BAM and VCF can be found at hts-specs. |
Imputed
WI.20210121.impute.isotype.vcf.gz WI.20210121.impute.isotype.vcf.gz.tbi |
| Reference Genome FASTA (WS276) | The reference genome build from Wormbase used for alignment and annotation. | 20210121_c_elegans_WS276.genome.fa |
| Gene models | Gene models were constructed using a combination of BRAKER (short-read) and StringTie + TransDecoder (long-read) followed by QC with AGAT using the reference genome CGC1. |
canonical_geneset.gtf.gz
annotations.gff3.gz current.geneIDs.txt.gz |
| Transposon Calls | We have performed transposon calling for C. elegans isotype reference strains as a part of Laricchia et al.. For C. briggsae and C. tropicalis, these data will be deposited as soon as they are generated. | 20210121_c_elegans_transposon_calls.bed is not included in this release |
| Genetic Map | A genetic map generated from a cross between N2 and CB4856 (Rockman & Kruglyak, 2009). | c_elegans_genetic_map.tsv is not included in this release |
| Tree | Tree generated using neighbour-joining algorithm as implemented in QuickTree in Newick and PDF format. |
All Strains
WI.20210121.hard-filter.min4.tree WI.20210121.hard-filter.min4.tree.pdf Isotype WI.20210121.hard-filter.isotype.min4.tree WI.20210121.hard-filter.isotype.min4.tree.pdf |
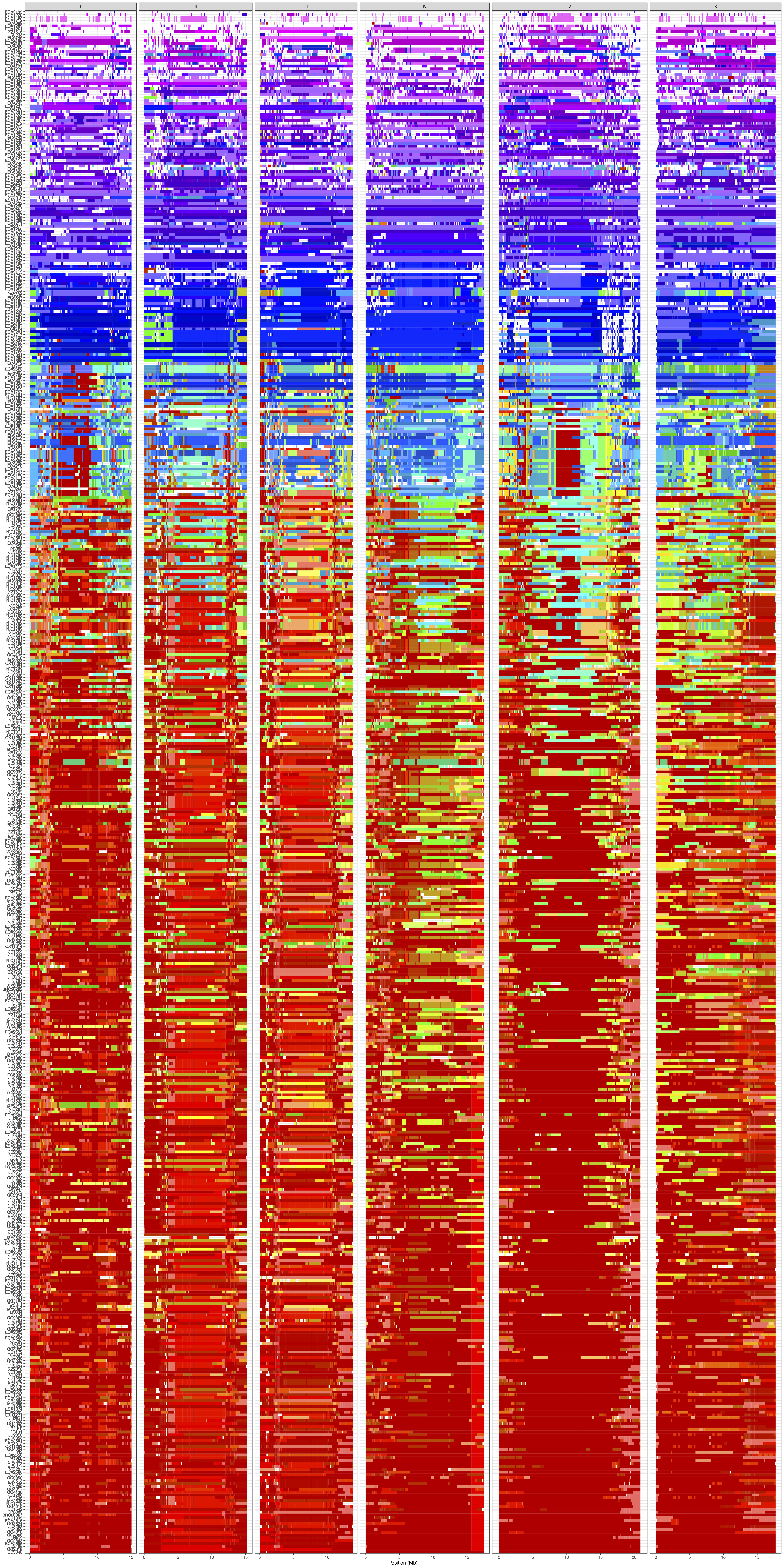
| Haplotypes | Haplotypes for isotypes were calculated and plotted as described in Lee et al. |
20210121_c_elegans_haplotype.png 20210121_c_elegans_haplotype.pdf |
| Sweep Haplotypes | The most frequent haplotype that covers at least 30% of the chromosome and is found on chromosome centers was determined and classified as a selective sweep. For more details of C. elegans selective sweeps, see Andersen et al. and Lee et al.. The plot shows red (swept), gray (non-swept), and white (not classified) regions. |
20210121_c_elegans_sweep.pdf 20210121_c_elegans_sweep_summary.tsv |
| Hyper-Divergent Regions | The hyper-divergent regions are characterized by higher-than-average density of small variants and large genomic spans where short sequence reads fail to align to the reference genome. For more information, see the FAQ. | 20210121_c_elegans_divergent_regions_strain.bed is not included in this release |
| Phenotype Trait Files | This gene expression trait was measured using short-read Illumina RNA-seq of mixed-stage populations of wild strains. Data are from Zhang et al. Nature Communications 2022. | 20231103_ZhangGeneExpression.tsv |
Methods / Pipelines
This tab links to the nextflow pipelines used to process wild isolate sequence data.

FASTQ QC and Trimming
andersenlab/trim-fq-nf -- (Latest d637d0b)
Adapters and low quality sequences were trimmed off of raw reads using fastp (0.20.0) and default parameters. Reads shorter than 20 bp after trimming were discarded.
Alignment
andersenlab/alignment-nf -- (1c96b4a)
Trimmed reads were aligned to C. elegans reference genome (project PRJNA13758 version WS276 from the Wormbase) using bwa mem BWA (0.7.17). Libraries of the same strain were merged together and indexed by sambamba (0.7.0). Duplicates were flagged with Picard (2.21.3).
Strains with less than 14x coverage were not included in the alignment report and subsequent analyses.
Variant Calling
andersenlab/wi-gatk -- (a84ba4f)
Variants for each strain were called using gatk HaplotypeCaller. After the initial variant calling, variants were combined and then recalled jointly using gatk GenomicsDBImport and gatk GenotypeGVCFs GATK (4.1.4.0).
The variants were further processed and filtered with custom-written scripts for heterozygous SNV polarization, GATK (4.1.4.0), and bcftools (1.10).
Heterozygous polarization and filtering thresholds were optimized for single nucleotide variants (SNVs).
Additionally, insertion or deletion (indel) variants less than 50 bp are more reliably called than indel variants greater than this size. In general, indel variants should be considered less reliable than SNVs.
Site-level filtering and annotation
andersenlab/post-gatk-nf -- (84d3a28)
-
Heterozygous SNV polarization: Because C. elegans is a selfing species, heterozygous SNV sites are most likely errors. Biallelic heterozygous SNVs were converted to homozygous REF or ALT if we had sufficient evidence for conversion. Only biallelic SNVs that are not on mitochondria DNA were included in this step. Specifically, the SNV was converted if the normalized Phred-scaled likelihoods (PL) met the following criteria (a smaller PL means more confidence). Any heterozygous SNVs that did not meet these criteria were left unchanged.
- If PL-ALT/PL-REF <= 0.5 and PL-ALT <= 200, convert to homozygous ALT
- If PL-REF/PL-ALT <= 0.5 and PL-REF <= 200, convert to homozygous REF
-
Soft filtering: Low quality sites were flagged but not modified or removed.
For the site-level soft filter, variant sites that meet the following conditions were flagged as PASS. These stats were computed across all samples for each site.
- Variant quality (QUAL) > 30 (this filter is very lenient, only three sites failed)
- Variant quality normalized by read depth (QD) > 20
- Strand bias of ALT calls: strand odds ratio (SOR) < 5
- Strand bias of ALT calls: Fisherstrand (FS) < 100
- Fraction of samples with missing genotype < 95%
- Fraction of samples with heterozygous genotype after heterozygous site polarization < 10%
For the sample-level soft filter, genotypes that meet the following filters were flagged as PASS for each site in each sample:
- Read depth (DP) > 5
- Site is not heterozygous
-
SnpEff Annotation: The predicted impact of each variant site was annotated with SnpEff (4.3.1t).
-
For the hard-filtered VCF, low quality sites were modified or removed using the following criteria.
- For the site-level hard filter, variant sites not flagged as PASS were removed.
- For the sample-level hard filter, genotypes not flagged as PASS were converted to missing (
./.), with the exception that heterozygous sites on mitochondria where kept unchanged.
After the steps above, sites that are invariant (
0/0or1/1across all samples, not counting missing./.) were removed. -
BCSQ Annotation: Variant impacts were then annotated using
bcftools csq, which takes into consideration nearby variants and annotates variant impacts based on haplotypes.
Determination of filter thresholds
We re-examined our filter thresholds for this release. A variant simulation pipeline was used as part of this process:
- Variant Simulations - andersenlab/variant-simulations-nf
Please see the filter optimization report for further details.
Isotype Assignment
andersenlab/concordance-nf -- (5160f9f)
Isotype groups contain strains that are likely identical to each other and were sampled from the same isolation locations. For any phenotypic assay, only the isotype reference strain needs to be scored. Users interested in individual strain genotypes can use the strain-level data.
Strains were grouped into isotypes using the following steps:
-
Using all high quality variants (only SNPs from the hard-filtered VCF) and
bcftools gtcheck, concordance for each pair of strains was calculated as a fraction of shared variants over the total variants in each pair. -
Strain pairs with concordance > 0.9997 were grouped into the same isotype group. The threshold 0.9997 was determined by:
- Examining the distribution of concordance scores.
- Capturing similarity between strains to minimize the number of strains that get assigned to multiple isotype groups.
- Agreement with the isotype groups in previous releases.
-
The following issues, which were rare, were resolved on a case-by-case basis:
- If one strain was assigned to multiple isotypes.
- If one isotype from previous releases matches to multiple new isotype groups.
- If one new isotype group contains strains from multiple isotypes from previous releases.
When issues arose, the pairwise concordance between all strains within an isotype were examined manually. Strains and isotypes may be re-assigned with the goal that strains within the same isotype group should have high concordance with each other, and strains from different isotype groups should have lower concordance.
Tree Generation
andersenlab/post-gatk-nf -- (84d3a28)
Trees were generated by converting the hard-filtered VCF to Phylip format using vcf2phylip (030b8d). Then, the Phylip format was converted to Stockholm format using Bioconvert (0.3.0), which was then used to construct a tree with QuickTree (2.5) using default settings. The trees were plotted with FigTree (1.4.4) rooting on the most diverse strain XZ1516.
Imputation
Imputation was not done for this release.