C. elegans releases
- Strains: 552
- WGS strains: 413
- Isotypes: 249
- Genome: WS258
Alignment Data
Wild isolate genomes are aligned and stored using the BAM format. BAMs are available in the latest release.
Methods
Information regarding alignment, variant calling, and annotation are available here.
Variant Data
We used samtools to identify single-nucleotide variant (SNV) sites as compared to the N2 reference genome. Variant data are provided as VCF or tab-delimited files.
VCF
VCFs generated from the variant calling pipeline are provided below.
- Soft-filter - Includes all variants and annotations. The QC status of variants is included.
- Hard-filter - Variants and genotypes that fail QC are removed.
- Imputed - An imputed dataset generated from the hard-filter VCF.
You can programmatically access specific regions of VCF files (rather then the entire file) from the command line:
Strain Data
Download Strain Data
Included Variants
Currently, we have performed variant calling across all wild isolates. We are working to add additional variant classes including insertion/deletion, structural, transposon, and additional variant classes.
Transposon Data
We have recently performed an analysis characterizing Transposon variation in C. elegans. The dataset will be further integrated with the site resources as time goes on. For now, the raw data are available below.
Download Transposon Data| # | Reference Strain | Isotype | Strains | Isotype BAM | Tab-delimited variants |
|---|
The following statistics were generated with bcftools stats. The soft-filtered VCF for this release has had records and genotypes annotated but no data has been removed. The hard-filtered VCF removes records and genotypes that have been annotated with filters.
The hard-filtered VCF has stripped records and genotypes that have had filters applied.
| Variable | Soft-Filtered | Hard-Filtered |
|---|---|---|
| Samples | 249 | 249 |
| SNVs | 2,434,802 | 1,865,541 |
| Ts | 1,361,672 | 1,041,676 |
| Tv | 1,153,388 | 823,865 |
| Ts/Tv | 1.18 | 1.26 |

Methods
Note: These methods operated on sequence data at the isotype level.
Software
Alignment
Sequences were aligned to WS245 using BWA (version 0.7.8-r455). Optical/PCR duplicates were marked with PICARD (version 1.111).
Variant Calling
SNV calling was performed using bcftools (version 1.3).
Filtering
Sites with greater than 10% missing or greater than 90% heterozygous calls across all isotypes were removed. Individual calls with the following parameters were removed:
- Depth of coverage (DP) <= 10
- Quality (QUAL) < 30
- Mapping Quality (MQ) < 40. Only applied to ALT calls.
- Number of high-quality non-reference bases (DV) / Depth of Coverage (DP) < 0.5. Applied only to ALT calls.
Annotation
Variants were annotated using SnpEff (version 4.1g) using the WS241 database.
Pipelines
The C. elegans Natural Diversity Resource has three git repos which contain the software used to run the site.
AndersenLab/cegwas
A set of functions to process phenotype data, perform GWAS, and perform post-mapping data processing for C. elegans.
AndersenLab/cegwas-worker
A python daemon that handles submitted mapping jobs from base. cegwas-worker Runs on Google Compute Engine.
AndersenLab/CeNDR
The software responsible for this website, which is run using Google App Engine.
Haplotypes
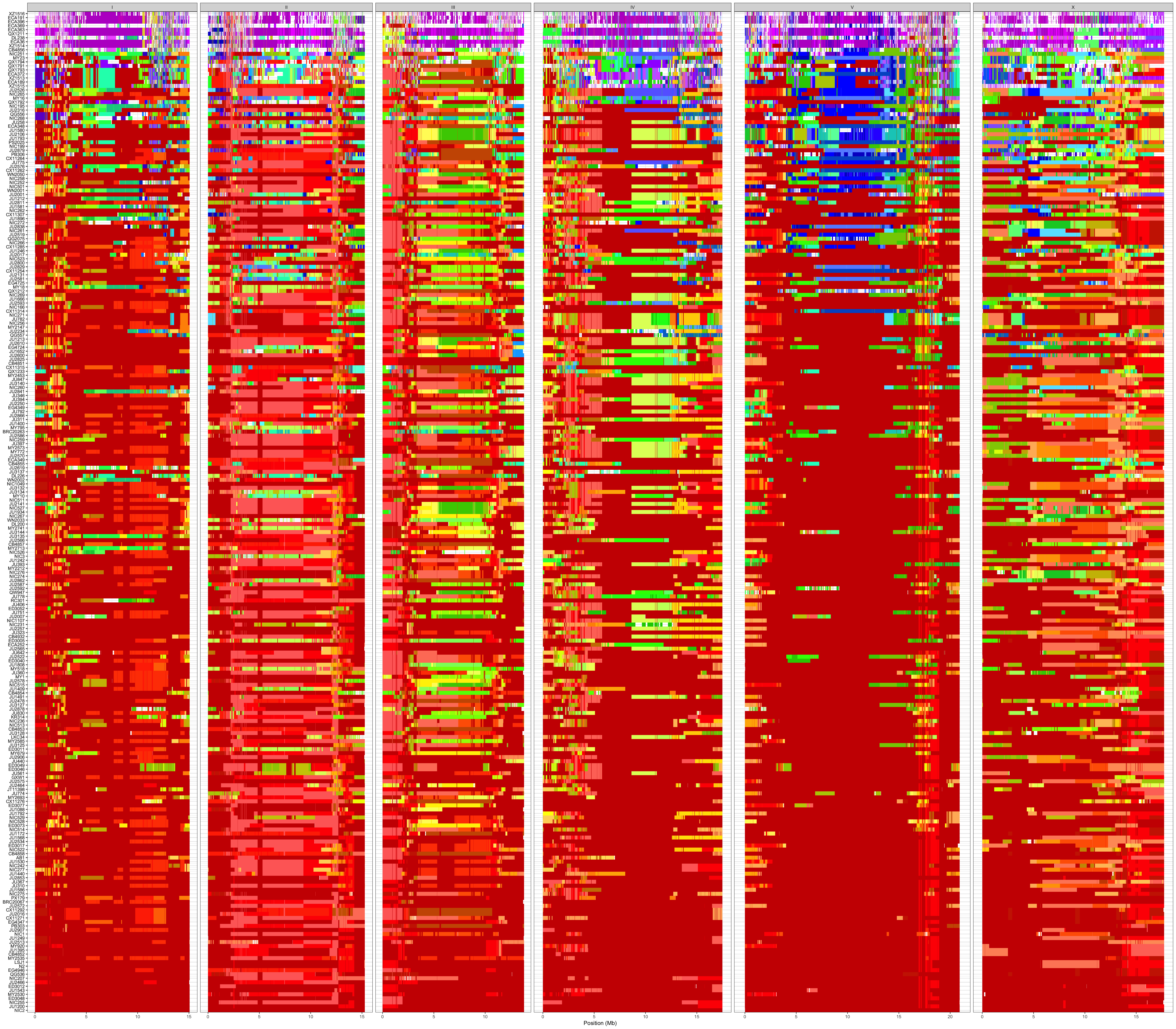 Click the above image to download the full version
Click the above image to download the full version