- How do I cite CaeNDR?
- What are hyper-divergent regions? How should I use variants that fall within these regions?
- Does CaeNDR have API access for computationally efficient data downloads?
- How much confidence do we have in the indel variants?
- How were the filter thresholds determined?
- What strains and data are available from CaeNDR?
- How are strains grouped by isotype?
- What are the differences in each tool? Why do some tools annotate variants and others don’t?
- What are compound variant annotations by CSQ?
- What are Grantham scores?
- What are BLOSUM62 scores?
- What is the percent protein metric?
- What is the divergent column of the annotation tool?
- How does CaeNDR decide on pricing?
How do I cite CaeNDR?
Please use the citation below.

CaeNDR, the Caenorhabditis Natural Diversity Resource
Please cite the efforts of researchers who have collected wild Caenorhabditis stains.
To cite the CaeNDR resource, please use the description below:
Timothy A Crombie, Ryan McKeown, Nicolas D Moya, Kathryn S Evans, Samuel J Widmayer, Vincent LaGrassa, Natalie Roman, Orzu Tursunova, Gaotian Zhang, Sophia B Gibson, Claire M Buchanan, Nicole M Roberto, Rodolfo Vieira, Robyn E Tanny, Erik C Andersen
(2023 Oct 19) Nucleic Acids Research
Or use this bibtex entry
@article{10.1093/nar/gkad887,
author = {Crombie, Timothy A and McKeown, Ryan and Moya, Nicolas D and Evans, Kathryn S and Widmayer, Samuel J and LaGrassa, Vincent and Roman, Natalie and Tursunova, Orzu and Zhang, Gaotian and Gibson, Sophia B and Buchanan, Claire M and Roberto, Nicole M and Vieira, Rodolfo and Tanny, Robyn E and Andersen, Erik C},
title = "{CaeNDR, the Caenorhabditis Natural Diversity Resource}",
journal = {Nucleic Acids Research},
volume = {52},
number = {D1},
pages = {D850-D858},
year = {2023},
month = {10},
abstract = "{Studies of model organisms have provided important insights into how natural genetic differences shape trait variation. These discoveries are driven by the growing availability of genomes and the expansive experimental toolkits afforded to researchers using these species. For example, Caenorhabditis elegans is increasingly being used to identify and measure the effects of natural genetic variants on traits using quantitative genetics. Since 2016, the C. elegans Natural Diversity Resource (CeNDR) has facilitated many of these studies by providing an archive of wild strains, genome-wide sequence and variant data for each strain, and a genome-wide association (GWA) mapping portal for the C. elegans community. Here, we present an updated platform, the Caenorhabditis Natural Diversity Resource (CaeNDR), that enables quantitative genetics and genomics studies across the three Caenorhabditis species: C. elegans, C. briggsae and C. tropicalis. The CaeNDR platform hosts several databases that are continually updated by the addition of new strains, whole-genome sequence data and annotated variants. Additionally, CaeNDR provides new interactive tools to explore natural variation and enable GWA mappings. All CaeNDR data and tools are accessible through a freely available web portal located at caendr.org.}",
issn = {0305-1048},
doi = {10.1093/nar/gkad887},
url = {https://doi.org/10.1093/nar/gkad887},
eprint = {https://academic.oup.com/nar/article-pdf/52/D1/D850/55039590/gkad887.pdf},
}
What are hyper-divergent regions? How should I use variants that fall within these regions?
Hyper-divergent regions are genomic intervals that contain sequences not found in the N2 reference strain. They were identified by high levels of variation and low coverage from read alignments. For a more full description, please read this paper. We highly recommend that you use the genome browser and view the BAM files for strains of interest. We also released a genomic view track to see where we have classified hyper-divergent regions. If you find that your region of interest overlaps with a hyper-divergent region, then we recommend taking any variants as preliminary. Long-read sequencing is required to identify the actual genomic sequences in this region.
Does CaeNDR have API access for computationally efficient data downloads?
Amazon Web Services (AWS) Open Data Project is making the CaeNDR data publicly available to the community free of charge. Public Data Sets on AWS provide a centralized repository of public data hosted on Amazon Simple Storage Service (Amazon S3). The data can be seamlessly accessed from AWS services such Amazon Elastic Compute Cloud (Amazon EC2) and Amazon Elastic MapReduce (Amazon EMR), which provide organizations with the highly scalable compute resources needed to take advantage of these large data collections. Researchers pay only for the additional AWS resources they need for further processing or analysis of the data. Learn more about Public Data Sets on AWS.
The latest CaeNDR Project data is publicly available in the CaeNDR Amazon S3 bucket.
We are working to implement an API to provide even more seamless data downloads.
How much confidence do we have in the indel variants?
GATK calls indel variants (1-50 bp) and short structural variants. The variant calling at these sites was not optimized and ran default parameters. These variants should be considered preliminary until confirmed by PCR or long-read sequencing.
How were the filter thresholds determined?
Optimal filter thresholds would faithfully separate real variant sites from non-variant sites. However, we had no way to know which variant sites were true or false using the experimental data. Therefore, we created simulated data with a "truth set" of variants artificially inserted into a BAM file. In this way, we know precisely the positions of true variants. After variant calling with the simulated BAM file, we looked at the various quality metrics and asked what thresholds of these metrics would best separate real variants from incorrectly called variants. We chose filter thresholds to maximize true positive rate and precision while minimizing the false positive rate. These filter thresholds were used in processing the wild isolate data.
See our filter optimization report for further details
What strains and data are available from CaeNDR?
The strains available on CaeNDR were sampled from all over the world by many members of the Caenorhabditis research community. CaeNDR endeavors to cryopreserve and whole-genome sequence all wild strains from C. briggsae, C. elegans, and C. tropicalis from all unique worldwide collection locations. Most times, independent strains collected from the same natural substrate are very genetically similar (or identical to the best level of determination by Illumina short-read sequencing SNV calling). Additionally, independent strains collected from nearby locations are often similar or identical. Because use of these strains in measurements of natural phenotypic diversity and in genome-wide association studies will cause significant bias, CaeNDR classifies strains that share high levels of genetic diversity as isotypes using whole-genome sequencing and variant calling. To remove this bias, we suggest one strain in each isotype, called the isotype reference strain. Strains and data on CaeNDR are focused on isotype reference strains. Users can request strains from the lab who originally identified the wild strains, but these strains are not genome-sequence verified and data on CaeNDR might not match strains from individual labs.
How are strains grouped by isotype?
In 2012, we published genome-wide variant data from reduced representation sequencing of approximately 10% of the C. elegans genome (RAD-seq). Using these data, we grouped strains into isotypes. We also found many strains that were mislabeled as wild isolates but were instead N2 derivatives, recombinants from laboratory experiments, and mutagenesis screen isolates (detailed in Strain Issues). These strains were not characterized further. For the isotypes, we chose one strain to be the isotype reference strain. This strain can be ordered through CaeNDR here.
After 2012, with advances in genome sequencing, we transitioned our sequencing to whole-genome short-read sequencing. All isotype reference strains were resequenced whole-genome. The other strains within an isotype were not, so we use the RAD-seq variant data to group isotypes for these strains.
What are the differences in each tool? Why do some tools annotate variants and others do not?
Annotations were obtained from ANNOVAR and VEP using default parameter settings, including the annotation of variants that are 5 kb upstream or downstream of a transcript for VEP (described in this paper and documented here), and 1 kb for ANNOVAR using their gene-based annotation mode (described in this paper). These parameters result in ANNOVAR and VEP annotating variants that fall in intergenic regions in addition to coding and regulatory regions.
VEP returns a qualitative impact score in addition to a predicted consequence. These impacts are HIGH, MODERATE, LOW, or MODIFIER based on the predicted consequence and if it is likely to disrupt protein function. For example, a stop_gained annotation would have an impact of HIGH, and a synonymous_variant annotation would have an impact of LOW.
ANNOVAR returns two consequence annotations, one that specifies where in a transcript a variant falls (exonic, splicing, UTR5, downstream, etc) and another that specifies the type of consequence that is predicted in that region (stoploss, nonsynonymous_SNV, frameshift_deletion, etc).
CSQ is haplotype-aware and does annotate intergenic variants. VEP and ANNOVAR can annotate variants in coding regions that CSQ does not annotate. This discrepancy in annotation is because of CSQ’s haplotype-aware functionality. If a deletion spans five base pairs at position 100 on chromosome I and then a single-nucleotide variant call at position 103 on chromosome I, CSQ will not annotate the variant at position 103 because it conflicts with the variant at position 100 and it cannot resolve the haplotype. Some annotations from CSQ will have an asterisk (*) before the predicted consequence. This asterisk indicates that the predicted consequence is downstream from a predicted nonsense (early stop) variant.
SnpEff is used for annotating MtDNA variants because it allows for the user to specify an MtDNA codon table, described here. For annotating MtDNA variants in Caenorhabditis, codon table 5 (invertebrate) is used.
VEP, SnpEff, and ANNOVAR annotate intergenic variants, but they differ in how they provide information on nearby transcripts. SnpEff reports the two transcripts that are flanking the variant position. VEP might report a transcript depending on the --distance threshold that is set, which defines how far upstream or downstream of a transcript a variant will be considered. ANNOVAR does not report any transcript resolution for intergenic variants. Because of these inconsistencies across tools, we have elected to remove transcript annotations for intergenic variants.
What are compound variant annotations by CSQ?
Bcftools’s variant annotation tool CSQ makes haplotype-aware variant annotations. If two or more variants are in the same haplotype and are observed in the same sample, then the combined change to the coding sequence of the genes is different than the single effects on the coding sequence. The creators provide some examples of these compound effects in their paper linked here.
An example of how a compound variant is reported is displayed here:
III,2371323,C,T,@2371328,@2371328,@2371328,
,NO,@2371328,@2371328,@2371328,N/A,N/A,N/A III,2371325,ATG,A,@2371328,@2371328,@2371328, ,NO,@2371328,@2371328,@2371328,N/A,N/A,N/A
The variants at position 2371323 and 2371325 are in the same haplotype as the variant at position 2371328, and together, they have a predicted consequence that is reported on the line where the 2371328 variant is reported and where the DNA change column is the ref>alt (@2371323) + ref>alt (@2371325) + ref>alt (@2371328):
III,2371328,C,CTA,stop_gained&inframe_altering,146AL>146*,2371323C>T+2371325ATG>A+2371328C>CTA,
,NO,H04J21.1.1,WBGene00019150,N/A,N/A,N/A,20.82
The reason CSQ annotations have a “DNA change” column but annotations from other tools do not is in order to differentiate predicted consequences that are from a compound annotation versus an individual variant annotation.
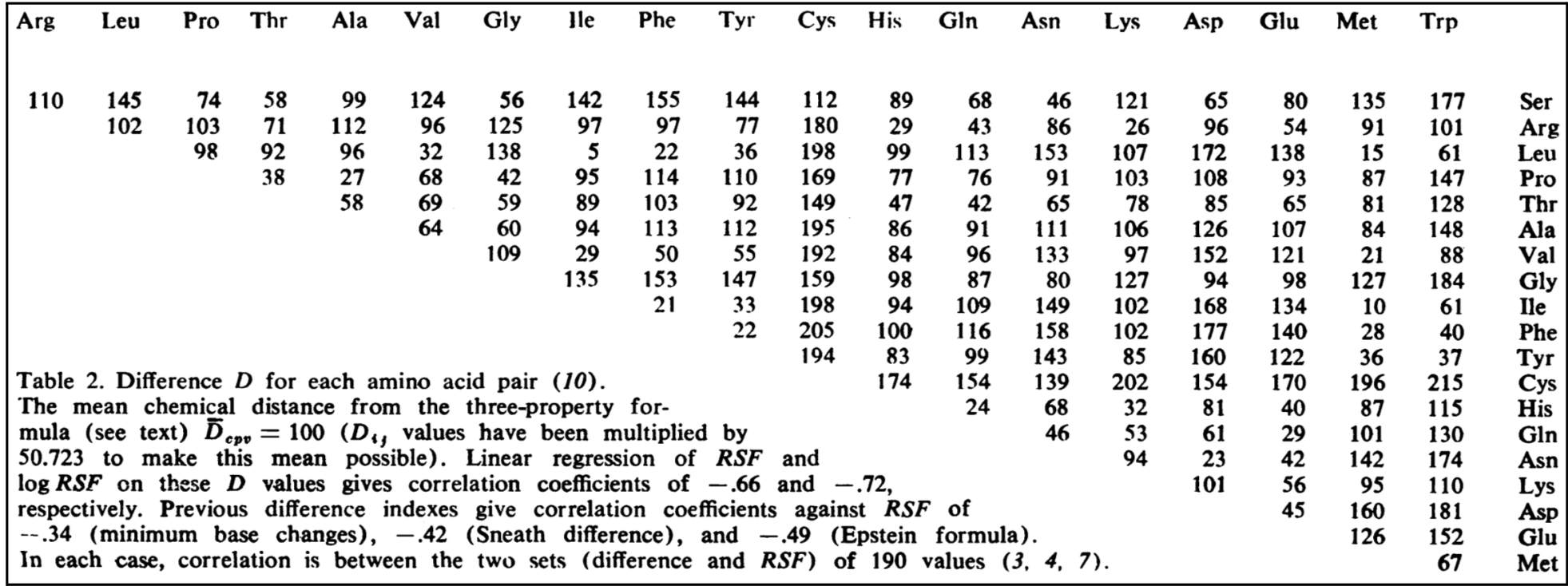
What are Grantham scores?
Grantham scores are a quantitative measure of how different an amino acid is from another based on combining properties of composition, polarity, and molecular volume and range from 5 to 215, described here. Amino acid pairs that have a high Grantham score are predicted to be more deleterious than amino acid pairs with a lower score. The Grantham matrix used for scoring is below:
What are BLOSUM62 scores?
BLOSUM62 (BLOcks SUbstitution Matrix) scores represent the log-odds of observing a given amino acid substitution (REF → ALT) compared to what would be expected by chance. Positive scores indicate amino acid substitution that are observed more frequently in evolutionarily related protein sequences, and negative scores indicate rare substitutions that can be deleterious to protein function. The BLOSUM62 matrix was derived from roughly 2,000 conserved regions ("blocks") across over 500 families of related proteins that share at least 62% sequence identity, described here. SnpEff does not have BLOSUM62 scores because the BLOSUM62 matrix was created based on nuclear proteins.

What is the percent protein metric?
The percent protein metric is a calculation of where in the protein a variant is present. The percent protein is calculated based on the nucleotide position of a variant in relation to the entire coding sequence, and accounts for Watson-Crick strandedness. All the coding regions for a transcript are summed, then the difference in the nucleotide position of a variant in relation to the position of where the first coding region begins is calculated and then divided by the total number of nucleotides in the coding region for a transcript.
What is the divergent column of the annotation?
Previous studies in the Andersen Laboratory have identified punctuated genomic regions that have extreme genetic variation in selfing Caenorhabditis species. These regions are referred to as hyper-divergent regions (HDRs). To learn more, please read this paper. HDRs are different for each wild strain. Therefore, we report if a variant call falls in an HDR for a given wild strain. If the divergent column has a value of YES, then the variant position is in an HDR for the strains that have the ALT allele at the variant position. Inversely, if the divergent column has a value of NO, then the variant position is possibly not in an HDR for the strains that have the ALT allele at the variant position. Notably, HDRs are identified differently in Caenorhabditis briggsae than in C. elegans and C. tropicalis. C. briggsae has higher levels of standing genetic variation than C. elegans and C. tropicalis, and its populations show structure with distinct relatedness groups. Some of these relatedness groups do not have a quality reference genome, and the range of genetic variation within each relatedness group varies, making parameter thresholds difficult for calling HDRs. The large range of genetic variation within C. briggsae and lack of representative reference genomes for some relatedness groups cause some HDRs to be falsely unannotated in some strains. We warn users that the HDR column could report a false negative NO value.
How does CaeNDR decide on pricing?
Prices for individual strains, diversity sets, and larger strain panels reflect the costs of strain cryopreservation, whole-genome sequencing, compute costs for data processing, web hosting costs, and data storage. Increases are evaluated every three years in response to these costs in consultation with members of the Advisory Committee.